小儿糖原贮积病Ⅳ型
糖原贮积病Ⅳ型(
glycogen storage disease type Ⅳ)又称支链淀粉病(amylopectinosis)、Andersen病(Andersens disease),是由淀粉1,4-1,6转葡萄糖苷酶缺陷所致。在病人的肝、肾、脾、肌肉及神经系统均有异常的糖原贮积,尤以肝细胞中贮积量最多。病儿在婴儿期即表现为生长障碍、肝
脾肿大、进行性门脉性纤维化导致
肝硬化、
腹水,多在早期死亡。寿命最长者为4年。检查肝、白细胞及成纤维细胞的分支酶可确定诊断。智能正常。本病无特殊治疗,仅作对症处理。有报告用皮质类固醇治疗可使病情暂时缓解。一例病儿用α-葡萄糖苷酶静脉注射4天,使肝脏的肝糖原含量从11%降到1%,但却没能改善临床过程。
 流行病学
流行病学
病因
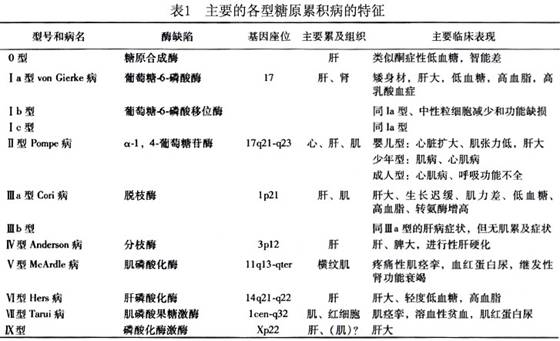
病因:本型是由于分支酶(brancher enzyme)缺陷所致,分支酶的编码基因(GBE1)位于3p12,根据现有资料分析,本病为常染色体隐性遗传病,但因患儿绝大多数为男性,故性连锁遗传的可能性尚不能除外。
发病机制
发病机制:分支酶又称为α-1,4-葡聚糖:α-1,4-葡聚糖-6-葡糖基转移酶(α-1,4-glucan-6-glycosyl transferase),是糖原合成途径中必需的酶。缺乏此酶使糖原合成时形成的直链增长,分支点锐减,糖原分子结构近似植物的支链淀粉而难溶于水。这种结构异常的糖原分子导致了肝脏严重的进行性损害,其机制尚不清楚。约半数本病患儿可同时有心肌、骨骼肌、中枢神经系统等损害,甚至有少数患者以肌肉、神经系统病变为主。因此,目前推测分支酶可能有两种从属于不同器官的同工酶,不同患者的临床表现取决于其酶缺陷的种类。这样,也可能解释了为什么这种结构异常的糖原分子,中仍然有少量分支点(1,6糖苷键)存在。
肝脏呈现结节性硬化,肝细胞排列不规则,纤维组织增生。肝细胞浆内无色或染色较浅的包涵体沉积,其边缘与胞浆分界明显,包涵体内呈玻璃样或网状结构,细胞核常偏于一旁。电镜检查和组织化学染色可显示异常结构的糖原。
临床表现
临床表现:患儿在出生后数月内常可无任何症状,而在3~15个月时逐渐出现肝、
脾大,腹部膨胀,消化道症状和体重不增等情况,并可能有肌张力低下,肌肉消瘦和萎缩、深腱反射消失等神经系症状。随着病情进展,
肝硬化和门脉高压征象逐渐明显,出现
腹水、腹壁静脉怒张和食管静脉曲张、
黄疸等。患儿甚易并发各种感染,常在3~4岁以内死于慢性肝功能衰竭。
分支酶缺陷的临床表现是多种多样的,近年已有少数经酶活性检测证实的成人病例报道,这些患者都是以神经、肌肉疾病症状为主。已知这类患者的外周神经组织和白细胞中的分支酶活力下降,随着资料的累积,将会有进一步认识。
并发症
并发症:体重不增,可发生
肝硬化和门脉高压,出现
腹水、腹壁静脉怒张和食管静脉曲张、
黄疸等,易并发各种感染,死于慢性肝功能衰竭。
实验室检查
实验室检查:本病常无低血糖表现,口服葡萄糖和
果糖耐量试验亦正常。胰高糖素或肾上腺素试验可使血糖轻度上升(0.8~1.3mmol/L),峰值常在30min时出现。
血清胆固醇轻度增高,血清转氨酶和碱性磷酸酶活力显著增高。血清蛋白质和
血氨等常随肝功能恶化而异常。酶活性检测可采用肝、肌组织或红、白细胞进行。
其他辅助检查
其他辅助检查:常规做X线胸片、B超、心电图和肌电图、脑电图检查。一般可见肝脏增大,脾脏增大,可发现腹水,肝硬化,食管静脉曲张等。肌电图、脑电图检查异常。
诊断
鉴别诊断
治疗
治疗:迄今为止,除一般支持治疗外,尚无有效治疗方法。已证实高蛋白、高脂肪、低碳水化合物饮食,胰高糖素和α-葡糖苷酶等对本病无效。对病损仅限于肝脏者,可考虑肝移植术。
预后
预后:本症预后差,多在早期死亡。
预防
预防:检测培养羊水细胞或绒毛细胞中的分支酶活力可供产前诊断。必要时终止妊娠。